


结直肠癌(Colorectal cancer, CRC)为癌症死亡率第二的疾病,其发病率一直处于上升趋势,可能与生活方式和环境因素的改变相关。近年来,生活方式和环境因素对肠道菌群影响的研究被不断报道,且发现了肠道菌群在CRC中的改变,但是功能方面的研究仍缺乏,也没有基于“多界”微生态的系统研究。上海交通大学医学院王慧教授与同济大学朱瑞新教授、中科院上海营养与健康研究所张国庆教授、复旦大学陈兴栋教授、中山大学附属第六医院朱立新教授团队合作,在Nature Microbiology(IF=30.964)上发表了题为“Multi-kingdom microbiota analyses identify bacterial–fungal interactions and biomarkers of colorectal cancer across cohorts”的研究成果。

文章总述
该研究通过对多中心队列的CRC患者的粪便样本进行宏基因组分析,绘制了来自7个国家(中国、法国、德国、奥地利、意大利、美国和日本)8个结直肠癌人群队列1368个样本(除中国之外的人群为网站获取公开数据)的“4K”微生态(细菌、真菌、古菌、病毒)图谱,揭示了与CRC相关的“四界肠道微生物”和功能的变化及彼此的可能互作机制,并建立了基于多界微生物的CRC诊断模型;最后也对肠道菌群介导的功能进行研究并通过qPCR对相关基因进行验证。
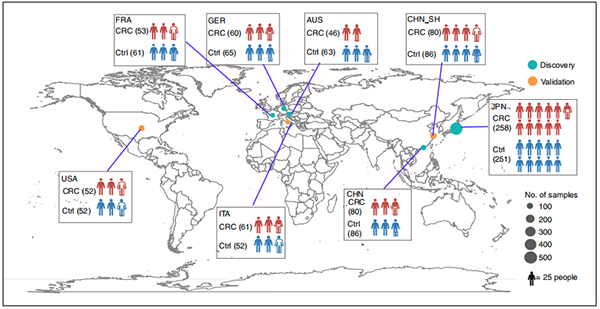
图1. 人群队列分布
结果
1.与CRC相关的菌变化
结合MMUPHin和PERMANOVA分析发现 ,CRC人群的α多样性显著降低;β多样性表明菌群的组成不仅与疾病有关,在不同人群间也有显著差异。对不同人群物种组成的MaAsline2分析发现与CRC相关各界菌的变化,同时也发现部分差异细菌(如Alistipes onderdonkii, Parvimonas micra和Gemella morbillorum)和差异真菌(如Aspergillus rambellii和Trichophyton mentagrophytes)稳定存在于5个发现数据中,也有部分古菌和病毒在不同数据集中存在明显差异。之后发现在CRC人群和对照人群中有显著差异的88个细菌、108个真菌、38个古菌和115个病毒,其中有48个已报道过的细菌在CRC中升高(如F.nucleatum,P. micra, Porphyromonas asaccharolytica, Desulfovibrio desulfuricans和Akkermansia muciniphila),有益的产乳酸菌在CRC中降低(Clostridium butyricum和Roseburia intestinalis等)。

图2. 微生物多样性和差异组成结果
进一步的机器学习算法选出与CRC相关的重要菌(27个细菌,20个真菌,20个古菌和21个病毒);建立的随机森林模型发现,细菌panel对CRC的诊断能力最强(平均AUROC=0.80),其次为真菌(平均AUROC=0.77),古菌(平均AUROC=0.74)和病毒(平均AUROC=0.73);为探讨基于各界panel建立的模型在不同数据集间的稳定性,又进行了cohort-to-cohort transfer和leave-one-cohort-out分析,进一步证实了建立模型的有效性。

图3. 基于单界微生态的差异菌和模型结果
随后又分别建立two-kingdom, three-kingdom和four-kingdom 微生物的multi-kingdom组合模型,发现multi-kingdom模型的诊断准确率优于single-kingdom模型,其中有细菌和真菌features的模型效果更好,并通过four-kingdom微生物组合模型确定了前20个最重要的features (13个细菌,5个真菌,1个古菌和1个病毒)。为进一步缩小核心菌群,通过随机森林确定了包含11种细菌、4种真菌和1种古菌在内的16个features的multi-kingdom模型,该模型在不同队列中均表现出较好的诊断效果(AUROC>0.82, 日本队列中AUROC=0.73),在德国队列中的AUROC高达0.92。此外,该模型也可用于CRC的早期诊断(AUROC=0.78)。
接着又用3个数据集(中国,奥地利和美国)对16个features的multi-kingdom模型进行验证,发现模型在中国和奥地利队列中的AUROC分别为0.88和0.81,而在美国队列中的AUROC为0.68,可能与该队列样本保存时间过长(>25年)有关。
为验证模型的特异性,又纳入IBD(inflammatory bowel disease),T2D(type 2 diabetes)和PD(Parkinson’s disease)三种疾病的数据集进行验证,结果发现模型的准确性大大降低,从而表明该模型在CRC诊断方面表现出有较高的特异性。
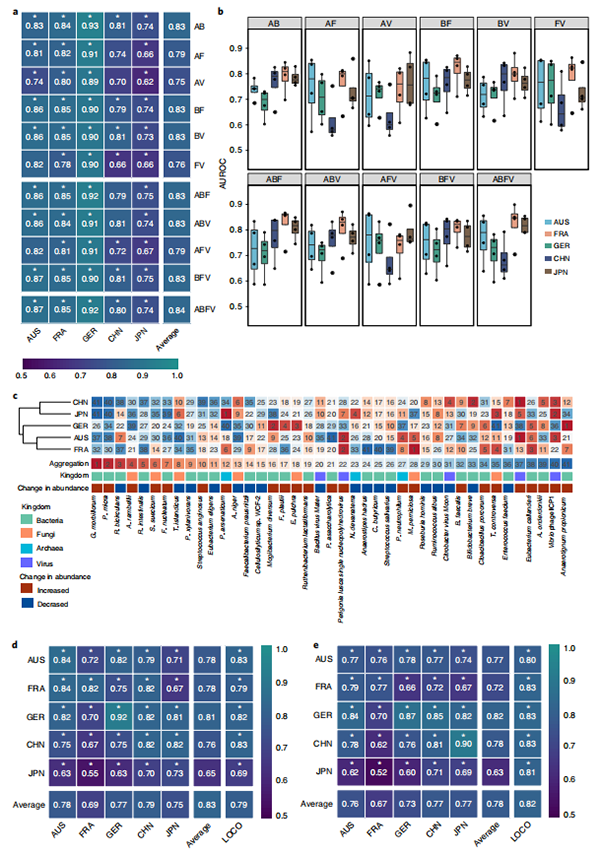
图4. Multi-kingdom模型结果
为进一步探究各菌间的相关性,基于各菌的丰度进行了co-abundance相关性分析,菌在CRC组中有更多的关联,其中除了kingdom内显著的关联外,还发现了kingdom间(特别是细菌-真菌)的显著关联。

图5. 相关性结果
2.微生物介导的功能变化和相关基因验证
为进一步探索微生物介导的功能变化,基于KEGG进行KO和pathway水平的功能分析,发现了1,053个差异KO基因和49个差异pathways,与碳水化合物代谢相关的pathways(如:丁酸,抗坏血酸和醛酸代谢)和与氨基酸代谢相关的pathways(D-精氨酸和D-鸟氨酸代谢)在CRC中升高,支链氨基酸和脂质代谢相关的pathways在CRC中减低;通过HAlla分析也发现了不同代谢通路与菌有一定的相关性。
对与D-精氨酸,D-鸟氨酸代谢相关基因oraE和oraS及与丁酸代谢相关的基因bdhA/B的PCR定量检测分析发现,在CRC中这几个基因显著升高。
该研究还进一步探索了multi-kingdom微生物之间的相互作用以及微生物介导的功能变化,发现了细菌-真菌相互作用通过上调D-精氨酸、D-鸟氨酸以及刺激丁酸代谢通路促进结直肠癌发病的机制。同时,该研究还基于功能基因构建了CRC诊断模型,也具有较高的准确率(平均AUC为0.86)。最后,基于EggNOG基因、KO基因和通路水平建立模型。有趣的是,基于EggNOG基因的诊断模型的准确率高达0.86,优于菌species水平建立的模型。

图6. 与CRC相关的功能变化和基于KO基因建立模型结果
总结
该研究是迄今为止样本量最大、最全面的基于宏基因组测序技术对CRC肠道菌群进行研究的多中心队列,首次绘制了全球CRC人群队列的“4K”微生态图谱,建立了基于多界组合的微生物组的CRC诊断模型,验证了由微生物介导功能相关基因的变化,为结直肠癌早期诊断和预后评估及潜在的治疗靶点提供了基于微生物及其功能标志物的新方法和新思路。
文献下载链接:
https://pan.baidu.com/s/1mJXsx8y5Qi5Vq_XF8yVHiw
提取码:0000









